GtoPdb is requesting financial support from commercial users. Please see our sustainability page for more information.
Voltage-gated sodium channels (NaV): Introduction

Introduction
Voltage-gated sodium channels are responsible for action potential initiation and propagation in excitable cells, including nerve, muscle, and neuroendocrine cell types [30,32]. They are also expressed at low levels in nonexcitable cells, where their physiological role is unclear [3]. Sodium channels are the founding members of the ion channel superfamily in terms of their discovery as a protein and determination of their amino acid sequence [62]. This article presents an introduction to their biochemical, molecular, and genetic properties, physiological roles, and pharmacological significance.
Sodium channel subunits
Sodium channels consist of a highly processed α subunit, which is approximately 260 kDa, associated with auxiliary β subunits of 33-39 kDa [5]. Sodium channels in the adult central nervous system (CNS) and heart contain a mixture of β1 - β4 subunits, while sodium channels in adult skeletal muscle have only the β1 subunit [4,33]. The pore-forming α subunit is sufficient for functional expression, but the kinetics and voltage-dependence of channel gating are modified by the β subunits and these auxiliary subunits are involved in channel localization and interaction with cell adhesion molecules, extracellular matrix and intracellular cytoskeleton. The α subunits are organised in four homologous domains (I–IV), which each contain six transmembrane alpha helices (S1–S6) and an additional pore loop located between the S5 and S6 segments (Figure 1; [5]). The pore loops line the outer entry to the pore while the S5 and S6 segments line the inner cavity and form activation gate at the inner exit from the pore. The S4 segments in each domain contain positively charged amino acid residues (usually arginine) at every third position. These residues serve as gating charges and move across the membrane in order to initiate channel activation in response to depolarization. The short intracellular loop connecting homologous domains III and IV serves as the inactivation gate, folding into the channel structure and blocking the pore from the inside during sustained depolarisation of the membrane.
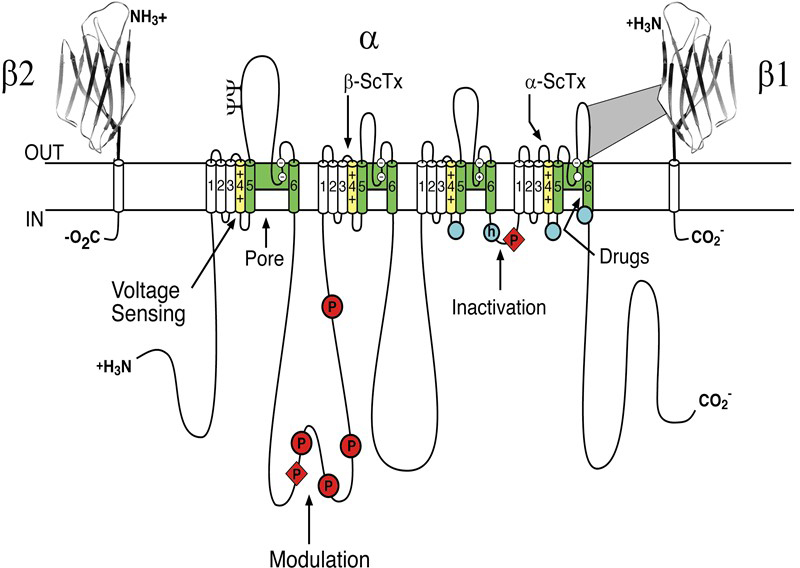
Figure 1. The primary structures of the subunits of the voltage-gated sodium channels. Cylinders represent probable α-helical segments. Bold lines represent the polypeptide chains of each subunit, with length approximately proportional to the number of amino acid residues in the brain sodium channel subtypes. The extracellular domains of the β1 and β2 subunits are shown as immunoglobulin-like folds. ψ, sites of probable N-linked glycosylation; P in red circles, sites of demonstrated protein phosphorylation by protein kinase A (circles) and protein kinase C (diamonds); green, pore-lining S5-P-S6 segments; white circles, the outer (EEDD) and inner (DEKA) rings of amino residues that form the ion selectivity filter and tetrodotoxin binding site; yellow, S4 voltage sensors; h in blue circle, inactivation particle in the inactivation gate loop; blue circles, sites implicated in forming the inactivation gate receptor. Sites of binding of α- and β-scorpion toxins and a site of interaction between α and β1 subunits are also shown. Tetrodotoxin is a specific blocker of the pore of sodium channels, whereas the α- and β-scorpion toxins block fast inactivation and enhance activation, respectively, and thereby generate persistent sodium current that causes depolarization block of nerve conduction. Click image for full size.
Sodium channel interacting proteins
Many proteins have been shown to interact with sodium channels transiently in the context of channel regulation, including several protein kinases and G proteins [7]. Some proteins interact more permanently in the context of subcellular targeting and localization, including cell adhesion molecules and cytoskeletal proteins [7]. In contrast to these interactions that are involved in targeting and regulation, a small number of proteins are thought to bind to sodium channels during their initial assembly and to serve as quasi-subunits that remain bound throughout the life of the channel molecule [17]. The ubiquitous calcium-dependent regulator calmodulin binds to a conserved site in the proximal C-terminal domain of many sodium channels and serves as a target for calcium regulation [23]. Fibroblast Growth Factor Homologous Factors (FHFs), which are intracellular regulators distantly related to Fibroblast Growth Factor, also form a tight complex with the proximal C-terminus of sodium channels and alter their function [37,42,56]. Mutations that disrupt interactions with FHFs cause spinocerebellar ataxia and cardiac arrhythmia, further emphasizing the importance of these interacting proteins [36,40].
Sodium channel structure at atomic resolution
Sodium channel architecture has been revealed in three-dimensions by determination of the crystal structure of bacterial sodium channels at high resolution (2.7Å) (Figure 2; [11,44,64], which has given new insight into the molecular basis for ion conductance and voltage-dependent activation. As viewed from the top, the bacterial sodium channel NavAb has a central pore surrounded by four pore-forming modules composed of S5 and S6 segments and the intervening pore loop (Figure 2A). Four voltage-sensing modules composed of S1-S4 segments are symmetrically located around the outer rim of the pore module (Figure 2A). The overall pore architecture has a large external vestibule, a narrow ion selectivity filter containing the amino acid residues shown to determine ion selectivity, a large central cavity that is lined by the S6 segments and is water filled, and an intracellular activation gate formed at the crossing of the S6 segments at the intracellular surface of the membrane (Figure 2B; [44]). The activation gate at the intracellular end of the pore is tightly closed in the NavAb structure (Figure 2B). The transmembrane architecture of NavAb shows that the adjacent subunits have swapped their functional domains such that each voltage-sensing module is most closely associated with the pore-forming module of its neighbour (Figure 2C), similar to voltage-gated potassium channels [39]. It is likely that this domain-swapped arrangement enforces concerted gating of the four subunits or domains of sodium and potassium channels. The NavAb ion selectivity filter has a high-field-strength site at its extracellular end (Figure 2D), formed by the side chains of four glutamate residues [4] which are highly conserved and are key determinants of ion selectivity in vertebrate sodium and calcium channels [28]. This outer site is followed by two ion coordination sites formed by backbone carbonyls (Figure 2D). These coordination sites are well designed to bind Na+ with up to four planar waters of hydration but would be much too large to bind Na+ directly.
Voltage-dependent activation of sodium channels requires transmembrane movement of three to four gating charges in each domain [1,31-32]. The S4 segment is in a transmembrane position in the voltage sensors of NavAb (Figure 2E; [44]). Three key landmarks in the voltage sensor are shown: the extracellular negative cluster (ENC), the hydrophobic constriction site (HCS), and the intracellular negative cluster (INC). In this activated state, the R1-R3 gating charges interact with the ENC and R4 interacts with the INC. Models of voltage sensor function predict that the S4 segment is retracted toward the intracellular side of the membrane by the electrostatic force of the resting membrane potential in the resting state, with the R2, R3, and R4 gating charges on the intracellular side of the HCS [6,60]. Depolarization then removes the electrostatic force pulling inward on the positive gating charges, allowing the S4 segment to move outward along a spiral path, exchange ion pair partners, and initiate the conformational change that opens the pore. This ‘sliding helix’ model of voltage sensor function has gained support from a broad consensus of investigators [54].
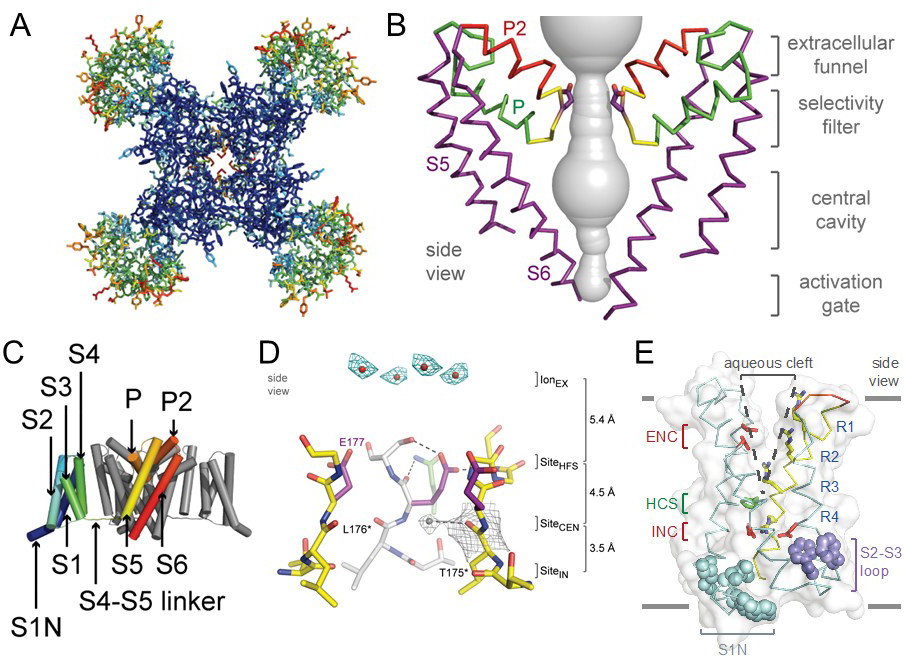
Figure 2. Structure of the bacterial sodium channel NavAb. A. Top view of NavAb channels colored according to crystallographic temperature factors of the main-chain (blue < 50Å [30] to red > 150Å [30]). The four pore modules in the center are rigid in the crystal structure and therefore are blue. The four voltage-sensing modules surround the pore and are more mobile, as illustrated by warmer colors. B. Architecture of the NavAb pore. Glu177 side-chains, purple; pore volume, grey. The P and P2 alpha helices that form the scaffold for the selectivity filter and outer vestibule are shown in green and red, respectively. C. Side view of NavAb. The structural components of one subunit are highlighted (1-6, transmembrane segments S1-S6). D. Side view of the selectivity filter. Glu177 (purple) interactions with Gln172, Ser178 and the backbone of Ser180 are shown in the far subunit; putative cations or water molecules (red spheres, IonEX). Electron-density around Leu176 (grey) and a bound water molecule are shown in gray. Na+-coordination sites: SiteHFS, SiteCEN and SiteIN. E. The structure of the voltage sensor. Side and of the voltage-sensing module of NavAb illustrating the extracellular negative charge-cluster (red, ENC), the intracellular negative charge-cluster (red, INC), hydrophobic constriction site (green, HCS), residues of the S1N helix (cyan) and phenylalanines of the S2-S3 loop (purple). S4 segment and gating charges (R1-R4) are in yellow. Insets. Examples of hydrogen bonding of gating-charges, dotted lines (<3.5Å). Click image for full size.
Sodium channel classification and nomenclature
A variety of different sodium channels have been identified by electrophysiological recording, biochemical purification, and cloning [26]. The sodium channels are members of the superfamily of ion channels that includes voltage-gated potassium and calcium channels [62]; however, unlike the different classes of potassium and calcium channels, the functional properties of the known sodium channels are relatively similar. Despite their similarity of function, the sodium channels were originally named in many different ways, with no consistent nomenclature for the various isoforms. To eliminate confusion resulting from the multiplicity of names, a standardised nomenclature was developed for voltage-gated sodium channels [27]. This nomenclature is based on that for voltage-gated potassium channels [13]. It utilises a numerical system to define subfamilies and sub-types based on similarities between the amino acid sequences of the channels. A comparable nomenclature has also been adopted for voltage-gated calcium channels ([19]; see the introductory article on Calcium Channels in this Database). In this nomenclature system, the name of an individual channel consists of the chemical symbol of the principal permeating ion (Na) with the principal physiological regulator (voltage) indicated as a subscript (NaV). The number following the subscript indicates the gene subfamily (currently only NaV1), and the number following the full point identifies the specific channel isoform (e.g. NaV1.1). This last number has been assigned according to the approximate order in which each gene was identified. Splice variants of each family member are identified by lower-case letters following the numbers (e.g. NaV1.1a).
The nine mammalian sodium channel isoforms that have been identified and functionally expressed are all greater than 50% identical in amino acid sequence in the transmembrane and extracellular domains, where the amino acid sequence is similar enough for clear alignment (Figure 3A). For potassium channels and calcium channels, all the members of distinct subfamilies are less than 50% identical to those of other subfamilies, and there is much closer sequence similarity within subfamilies [13,19]. The sodium channel sequences vary more continuously, without defining separate subfamilies. By this criterion, all of the nine sodium channel isoforms may be considered members of one protein family.
Sodium channel genes
To test this hypothesis more critically, the nine sodium channel amino acid sequences were aligned and compared for relatedness using a maximum parsimony procedure that measured their evolutionary distance by calculating the number of nucleotide changes required for the change in codon at each position (Figure 3B). The resulting phylogenetic tree is consistent with designation of these sodium channels as a single family. NaV1.1, NaV1.2, NaV1.3, and NaV1.7 are the most closely related group by this analysis. All four of these sodium channels are highly tetrodotoxin-sensitive and are broadly expressed in neurones. Their genes are all located on human chromosome 2q23-24, consistent with a common evolutionary origin. NaV1.5, NaV1.8, and NaV1.9 are also closely related (Figure 3B), and their amino acid sequences are greater than 64% identical to those of the four sodium channels encoded on chromosome 2. These sodium channels are tetrodotoxin-resistant to varying degrees due to changes in amino acid sequence at a single position in domain I, and they are highly expressed in heart and dorsal root ganglion (DRG) neurons [5,22,50]. Their genes are located on human chromosome 3p21-24, consistent with a common evolutionary origin. The isoforms NaV1.4, expressed primarily in skeletal muscle, and NaV1.6, expressed primarily in the central nervous system, are set apart from these other two closely related groups of sodium channel genes (Figure 3B). Although their amino acid sequences are greater than 84% identical to the group of sodium channels whose genes are located on chromosome 2 (Figure 3A), their phylogenetic relationship is much more distant when analyzed by parsimony comparison (Figure 3B). This distant evolutionary relationship is consistent with the location of the genes encoding these two sodium channels on chromosomes 17q23-25 and 12q13, respectively. The chromosome segments carrying the sodium channel genes are paralogous segments that contain many sets of related genes, including the homeobox (HOX) gene clusters. These segments were generated by whole genome duplication events during early vertebrate evolution [45]. The comparisons of amino acid sequence identity and phylogenetic and chromosomal relationships lead to the conclusion that all nine members of the sodium channel family that have been functionally expressed are members of a single family of proteins and have arisen from gene duplications and chromosomal rearrangements relatively recently in evolution. These results contrast with those for potassium channels and calcium channels, for which distinct gene families have arisen earlier in evolution and have been maintained as separate families to the present [13,19].
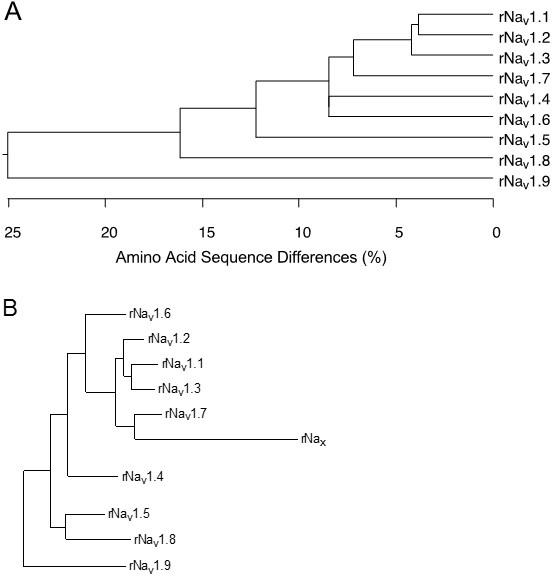
Figure 3. Amino acid sequence similarity and phylogenetic relationships of voltage-gated sodium channel α subunits. A. Comparison of amino acid identity for rat sodium channels Nav1.1 - Nav1.9. The comparison was performed with Megalign in the program DNAStar (utilizing the Clustal method) for the four domains and the cytoplasmic linker connecting domains III and IV. B. Phylogenetic relationships by maximum parsimony analysis of rat sodium channel sequences Nav1.1-Nav1.9 and Nax. To perform the analysis, the amino acid sequences for all isoforms were aligned using Clustal W. The amino acid sequences in the alignments were then replaced with the published nucleotide sequences, and the nucleotide sequence alignments were subjected to analysis using the program PAUP*. Divergent portions of the terminal regions and the cytoplasmic loops between domains I-II and II-III were excluded from the PAUP* analysis. The tree was rooted by including the invertebrate sodium channel sequences during the generation of the tree, although these sequences are not shown in the figure. Click image for full size.
In addition to these nine sodium channels that have been functionally expressed, closely related sodium channel-like proteins (Nax) have been cloned from mouse, rat, and human. They are approximately 50% identical to the NaV1 subfamily of channels but more than 80% identical to each other. They have significant amino acid sequence differences in the voltage sensors, inactivation gate, and pore region that are critical for channel function and have previously been proposed as a distinct subfamily [25]. They are closely related phylogenetically to the group of sodium channels on human chromosome 2q23-24, where their gene is also located [27]. These atypical sodium channel-like proteins are expressed in uterus, smooth muscle, astrocytes, and neurones in the hypothalamus and peripheral nervous system (PNS). These channels are not voltage-gated, but they are sodium selective [41]. Their activity is regulated by extracellular sodium concentration, and they are implicated in control of extracellular sodium concentration via glial cells in the subfornical organ in the hypothalamus [41]. They are also expressed in neurons, where their function remains uncertain.
Four auxiliary subunits of sodium channels have been defined thus far: NaVβ1, NaVβ2, NaVβ3, and NaVβ4 [5,33,63]. In the event that additional subunits are identified, we propose that the nomenclature should be comparable to that for the auxiliary subunits of calcium channels [19].
Sodium channel molecular pharmacology
All of the pharmacological agents that act on sodium channels have receptor sites on the α subunits. At least six distinct receptor sites for neurotoxins and one receptor site for local anesthetics and related drugs have been identified ([12], Table 1). Neurotoxin receptor site 1 binds the nonpeptide pore blockers tetrodotoxin (TTX) and saxitoxin and the peptide pore blocker μ-conotoxin [5,22,52]. The receptor sites for these toxins are formed by amino acid residues in the pore loops and immediately on the extracellular side of the pore loops at the outer end of the pore. Tetrodotoxin is noteworthy as the toxin of the Japanese puffer fish (‘fugu’), an edible delicacy, and saxitoxin is the toxin produced by some dinoflaggelates that cause red tides in cold ocean waters and can cause paralytic shellfish poisoning. Neurotoxin receptor site 2 binds a family of lipid-soluble toxins, including batrachotoxin, veratridine, aconitine, and grayanotoxin, which enhance activation of sodium channels. Photoaffinity labeling and mutagenesis studies implicate transmembrane segments IS6 and IVS6 in the receptor site for batrachotoxin [12]. Neurotoxin receptor site 3 binds the α-scorpion toxins and sea anemone toxins, which slow the coupling of sodium channel activation to inactivation. These peptide toxins bind to a complex receptor site that includes the S3-S4 loop at the outer end of the S4 segment in domain IV [12]. Neurotoxin receptor site 4 binds the β-scorpion toxins, which enhance activation of the channels. The receptor site for the β-scorpion toxins includes the S3-S4 loop at the extracellular end of the voltage-sensing S4 segments in domain II [12]. Neurotoxin receptor site 5 binds the complex polyether toxins brevetoxin and ciguatoxin, which are made by dinoflagellates and cause toxic red tides in warm ocean waters [12]. Transmembrane segments IS6 and IVS5 are implicated in brevetoxin binding from photoaffinity labeling studies [12]. Neurotoxin receptor site 6 binds δ-conotoxins, which slow the rate of inactivation like the α-scorpion toxins. The location of neurotoxin receptor site 6 is unknown. Finally, the local anesthetics and related antiepileptic and antiarrhythmic drugs bind to overlapping receptor sites located in the inner cavity of the pore of the sodium channel [5]. Amino acid residues in the S6 segments from at least three of the four domains contribute to this complex drug receptor site, with the IVS6 segment playing the dominant role.
TABLE 1: Receptor sites on sodium channels
| |||||||||||||||||||||||||||||||||||||||||||||||||||
Structure of the drug receptor site in sodium channels
Sodium channels are blocked by drugs used clinically as local anesthetics, antiarrhythmics, and antiepileptics. Site-directed mutagenesis studies revealed the receptor site for local anesthetics and related drugs, which is formed by amino acid residues in the S6 segments in domains I, III, and IV (Figure 4A) [47-48,57,59,61]. These drugs bind to a common receptor site in the pore and impede ion permeation. The amino acid residues that form the receptor sites for sodium channel blockers line the inner surface of the S6 segments and create a three-dimensional drug receptor site whose occupancy would block the pore (Figure 4B, C) [2,5,10,44,47,53,61]. Access to this receptor site by large or hydrophilic drugs would require opening of the intracellular activation gate, which is tightly closed (Figure 4B). This tight closure of the activation gate provides a structural basis for use-dependent block of sodium channels by local anaesthetics and related drugs [29], as they would bind much more rapidly when the channel is frequently opened. Remarkably, as predicted by the modulated receptor hypothesis [29], fenestrations lead from the lipid phase of the membrane sideways into the drug receptor site, providing a specific hydrophobic access pathway for binding of small hydrophobic drugs in the resting state of the channel (Figure 3B, C, pore portals) [44]. Access to the drug binding site in NavAb channels is controlled by the side chain of a single amino acid residue, Phe203 (Figure 4B, C) [44], which is homologous to amino acid residues identified in previous structure-function studies that control drug access and egress from the local anesthetic receptor site in mammalian cardiac and brain sodium channels [46-47]. The fenestrations change size and shape in the inactivated state of NavAb [43], providing a potential mechanism for preferential state-dependent binding of local anaesthetics and related drugs to inactivated sodium channels, as predicted by the modulated receptor hypothesis.
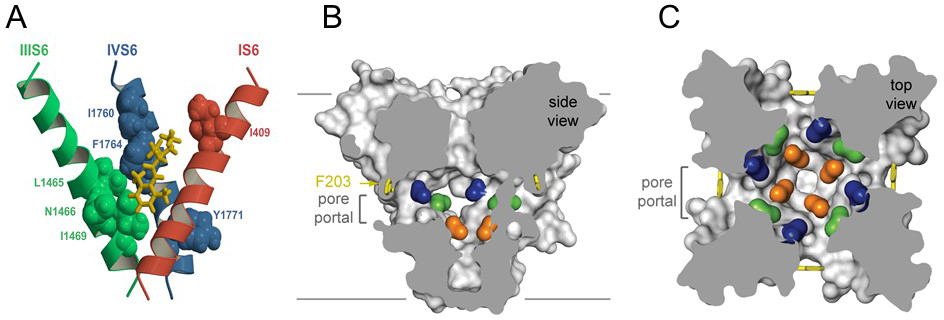
Figure 4. Drug binding site and access pathways in the central cavity of sodium channels. A. A model of the binding site for local anaesthetics and related antiepileptic and antiarrhythmic drugs in the IS6, IIIS6, and IVS6 transmembrane segments of a mammalian sodium channel. Amino acid side chains involved in drug binding are shown in spacefilling format. Yellow, bound etidocaine. B. Side-view through the pore module in the structure of NavAb illustrating fenestrations (portals) and hydrophobic access to central cavity. Phe203 side-chains, yellow sticks. Surface representations of NavAb residues aligning with those implicated in drug binding and block, Thr206, blue; Met209, green; Val213, orange. Membrane boundaries, gray lines. C. Top-view sectioned below the selectivity filter, colored as in A. Click image for full size.
Sodium channelopathies
A surprisingly large number of genetic diseases are caused by mutations of sodium channels, including inherited forms of periodic paralysis, cardiac arrhythmia, epilepsy, migraine, peripheral neuropathy, and chronic pain [9,14,18,20-21,24,35,38,55]. In most cases, these are genetically dominant diseases in which the mutations cause a gain-of-function effect at both the molecular and cellular levels (but see Dravet Syndrome below for an exception) and the resulting hyperexcitability leads to the symptoms of the disease [9,16,34-35,55]. However, in the rare recessive pain disorder Congenital Indifference to Pain, loss-of-function mutations in both alleles of the gene encoding NaV1.7 channels cause complete loss of pain sensation [15].
Unexpected gain-of-function mechanisms cause two of these sodium channelopathies. In Hypokalemic Periodic Paralysis, mutations in the gating charges of the skeletal muscle sodium channel NaV1.4 cause a leak of ions through the voltage sensor, which depolarizes muscle fibers, causes sodium leak, and leads to the symptoms of the disease [51]. In Dravet Syndrome, loss-of-function mutations in NaV1.1 channels selectively reduces action potential firing in GABAergic inhibitory interneurons, which leads to gain of function by disinhibition of neural circuits and causes the multifaceted phenotypes of this intractable childhood epilepsy syndrome [8].
As these examples illustrate, sodium channelopathies can be caused by either gain-of-function or loss-of-function at the molecular level of the channel. Moreover, some sodium channel mutations that are gain-of-function at the molecular level can have different or even opposing effects within different cell backgrounds. For example, a single sodium channel mutation can produce hyperexcitability in some classes of neurons while producing hypoexcitability in other types of neurons. This appears to be a consequence of the presence, in different types of neurons, of different levels of expression of other ion channels [49].
Sequencing of sodium channel genes is now available as a clinical test. In interpreting the results of sequencing of sodium channel genes, it is important to recognize that some variants of sodium channel genes, particularly ones in regions of the channel protein that are not membrane-spanning, are functionally silent and clinically insignificant. It has been recommended that mutations of sodium channel genes be considered as definitively pathogenic only when a clear effect on channel and/or cellular function has been demonstrated or when the variant has been shown to be present in multiple affected family members in a multi-generational kindred [58].
References
1. Armstrong CM, Bezanilla F. (1973) Currents related to movement of the gating particles of the sodium channels. Nature, 242 (5398): 459-61. [PMID:4700900]
2. Bagnéris C, DeCaen PG, Naylor CE, Pryde DC, Nobeli I, Clapham DE, Wallace BA. (2014) Prokaryotic NavMs channel as a structural and functional model for eukaryotic sodium channel antagonism. Proc Natl Acad Sci USA, 111 (23): 8428-33. [PMID:24850863]
3. Black JA, Waxman SG. (2013) Noncanonical roles of voltage-gated sodium channels. Neuron, 80 (2): 280-91. [PMID:24139034]
4. Brackenbury WJ, Isom LL. (2011) Na Channel β Subunits: Overachievers of the Ion Channel Family. Front Pharmacol, 2: 53. [PMID:22007171]
5. Catterall WA. (2000) From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron, 26 (1): 13-25. [PMID:10798388]
6. Catterall WA. (2010) Ion channel voltage sensors: structure, function, and pathophysiology. Neuron, 67 (6): 915-28. [PMID:20869590]
7. Catterall WA. (2010) Signaling complexes of voltage-gated sodium and calcium channels. Neurosci Lett, 486 (2): 107-16. [PMID:20816922]
8. Catterall WA. (2014) Sodium channels, inherited epilepsy, and antiepileptic drugs. Annu Rev Pharmacol Toxicol, 54: 317-38. [PMID:24392695]
9. Catterall WA, Dib-Hajj S, Meisler MH, Pietrobon D. (2008) Inherited neuronal ion channelopathies: new windows on complex neurological diseases. J Neurosci, 28 (46): 11768-77. [PMID:19005038]
10. Catterall WA, Swanson TM. (2015) Structural basis for pharmacology of voltage-gated sodium and calcium channels. Mol Pharmacol, 88 (1): 141-50. [PMID:25848093]
11. Catterall WA, Zheng N. (2015) Deciphering voltage-gated Na(+) and Ca(2+) channels by studying prokaryotic ancestors. Trends Biochem Sci, 40 (9): 526-34. [PMID:26254514]
12. Cestèle S, Catterall WA. (2000) Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie, 82 (9-10): 883-92. [PMID:11086218]
13. Chandy KG, Gutman GA. (1993) Nomenclature for mammalian potassium channel genes. Trends Pharmacol Sci, 14 (12): 434. [PMID:8122319]
14. Clancy CE, Kass RS. (2002) Defective cardiac ion channels: from mutations to clinical syndromes. J Clin Invest, 110 (8): 1075-7. [PMID:12393842]
15. Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y et al.. (2006) An SCN9A channelopathy causes congenital inability to experience pain. Nature, 444 (7121): 894-8. [PMID:17167479]
16. Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. (2007) From genes to pain: Na v 1.7 and human pain disorders. Trends Neurosci, 30 (11): 555-63. [PMID:17950472]
17. Dib-Hajj SD, Waxman SG. (2010) Isoform-specific and pan-channel partners regulate trafficking and plasma membrane stability; and alter sodium channel gating properties. Neurosci Lett, 486 (2): 84-91. [PMID:20817075]
18. Dib-Hajj SD, Yang Y, Black JA, Waxman SG. (2013) The Na(V)1.7 sodium channel: from molecule to man. Nat Rev Neurosci, 14 (1): 49-62. [PMID:23232607]
19. Ertel EA, Campbell KP, Harpold MM, Hofmann F, Mori Y, Perez-Reyes E, Schwartz A, Snutch TP, Tanabe T, Birnbaumer L, Tsien RW, Catterall WA. (2000) Nomenclature of voltage-gated calcium channels. Neuron, 25 (3): 533-5. [PMID:10774722]
20. Escayg A, Goldin AL. (2010) Sodium channel SCN1A and epilepsy: mutations and mechanisms. Epilepsia, 51 (9): 1650-8. [PMID:20831750]
21. Faber CG, Hoeijmakers JG, Ahn HS, Cheng X, Han C, Choi JS, Estacion M, Lauria G, Vanhoutte EK, Gerrits MM et al.. (2012) Gain of function Naν1.7 mutations in idiopathic small fiber neuropathy. Ann Neurol, 71 (1): 26-39. [PMID:21698661]
22. Fozzard HA, Hanck DA. (1996) Structure and function of voltage-dependent sodium channels: comparison of brain II and cardiac isoforms. Physiol Rev, 76 (3): 887-926. [PMID:8757791]
23. Gabelli SB, Boto A, Kuhns VH, Bianchet MA, Farinelli F, Aripirala S, Yoder J, Jakoncic J, Tomaselli GF, Amzel LM. (2014) Regulation of the NaV1.5 cytoplasmic domain by calmodulin. Nat Commun, 5: 5126. [PMID:25370050]
24. George AL. (2005) Inherited disorders of voltage-gated sodium channels. J Clin Invest, 115 (8): 1990-9. [PMID:16075039]
25. George Jr AL, Knittle TJ, Tamkun MM. (1992) Molecular cloning of an atypical voltage-gated sodium channel expressed in human heart and uterus: evidence for a distinct gene family. Proc Natl Acad Sci USA, 89 (11): 4893-7. [PMID:1317577]
26. Goldin AL. (2001) Resurgence of sodium channel research. Annu Rev Physiol, 63: 871-94. [PMID:11181979]
27. Goldin AL, Barchi RL, Caldwell JH, Hofmann F, Howe JR, Hunter JC, Kallen RG, Mandel G, Meisler MH, Netter YB et al.. (2000) Nomenclature of voltage-gated sodium channels. Neuron, 28 (2): 365-8. [PMID:11144347]
28. Heinemann SH, Terlau H, Stühmer W, Imoto K, Numa S. (1992) Calcium channel characteristics conferred on the sodium channel by single mutations. Nature, 356 (6368): 441-3. [PMID:1313551]
29. Hille B. (1977) Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J Gen Physiol, 69 (4): 497-515. [PMID:300786]
30. Hille B. (2001) Ionic Channels of Excitable Membranes, 3rd Ed. In (Sinauer Associates Inc.) .
31. Hirschberg B, Rovner A, Lieberman M, Patlak J. (1995) Transfer of twelve charges is needed to open skeletal muscle Na+ channels. J Gen Physiol, 106 (6): 1053-68. [PMID:8786350]
32. HODGKIN AL, HUXLEY AF. (1952) A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol (Lond.), 117 (4): 500-44. [PMID:12991237]
33. Isom LL. (2001) Sodium channel beta subunits: anything but auxiliary. Neuroscientist, 7 (1): 42-54. [PMID:11486343]
34. Jurkat-Rott K, Lehmann-Horn F. (2006) Paroxysmal muscle weakness: the familial periodic paralyses. J Neurol, 253 (11): 1391-8. [PMID:17139526]
35. Keating MT, Sanguinetti MC. (2001) Molecular and cellular mechanisms of cardiac arrhythmias. Cell, 104 (4): 569-80. [PMID:11239413]
36. Laezza F, Gerber BR, Lou JY, Kozel MA, Hartman H, Craig AM, Ornitz DM, Nerbonne JM. (2007) The FGF14(F145S) mutation disrupts the interaction of FGF14 with voltage-gated Na+ channels and impairs neuronal excitability. J Neurosci, 27 (44): 12033-44. [PMID:17978045]
37. Laezza F, Lampert A, Kozel MA, Gerber BR, Rush AM, Nerbonne JM, Waxman SG, Dib-Hajj SD, Ornitz DM. (2009) FGF14 N-terminal splice variants differentially modulate Nav1.2 and Nav1.6-encoded sodium channels. Mol Cell Neurosci, 42 (2): 90-101. [PMID:19465131]
38. Lehmann-Horn F, Jurkat-Rott K. (1999) Voltage-gated ion channels and hereditary disease. Physiol Rev, 79 (4): 1317-72. [PMID:10508236]
39. Long SB, Tao X, Campbell EB, MacKinnon R. (2007) Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature, 450 (7168): 376-82. [PMID:18004376]
40. Musa H, Kline CF, Sturm AC, Murphy N, Adelman S, Wang C, Yan H, Johnson BL, Csepe TA, Kilic A et al.. (2015) SCN5A variant that blocks fibroblast growth factor homologous factor regulation causes human arrhythmia. Proc Natl Acad Sci USA, 112 (40): 12528-33. [PMID:26392562]
41. Noda M, Hiyama TY. (2015) The Na(x) Channel: What It Is and What It Does. Neuroscientist, 21 (4): 399-412. [PMID:24962095]
42. Pablo JL, Pitt GS. (2016) Fibroblast Growth Factor Homologous Factors: New Roles in Neuronal Health and Disease. Neuroscientist, 22 (1): 19-25. [PMID:25492945]
43. Payandeh J, Gamal El-Din TM, Scheuer T, Zheng N, Catterall WA. (2012) Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature, 486 (7401): 135-9. [PMID:22678296]
44. Payandeh J, Scheuer T, Zheng N, Catterall WA. (2011) The crystal structure of a voltage-gated sodium channel. Nature, 475 (7356): 353-8. [PMID:21743477]
45. Plummer NW, Meisler MH. (1999) Evolution and diversity of mammalian sodium channel genes. Genomics, 57 (2): 323-31. [PMID:10198179]
46. Qu Y, Rogers J, Tanada T, Scheuer T, Catterall WA. (1995) Molecular determinants of drug access to the receptor site for antiarrhythmic drugs in the cardiac Na+ channel. Proc Natl Acad Sci USA, 92 (25): 11839-43. [PMID:8524860]
47. Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. (1994) Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science, 265 (5179): 1724-8. [PMID:8085162]
48. Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. (1996) Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of voltage-gated Na+ channels. Proc Natl Acad Sci USA, 93 (17): 9270-5. [PMID:8799190]
49. Rush AM, Dib-Hajj SD, Liu S, Cummins TR, Black JA, Waxman SG. (2006) A single sodium channel mutation produces hyper- or hypoexcitability in different types of neurons. Proc Natl Acad Sci USA, 103 (21): 8245-50. [PMID:16702558]
50. Sivilotti L, Okuse K, Akopian AN, Moss S, Wood JN. (1997) A single serine residue confers tetrodotoxin insensitivity on the rat sensory-neuron-specific sodium channel SNS. FEBS Lett, 409 (1): 49-52. [PMID:9199502]
51. Sokolov S, Scheuer T, Catterall WA. (2007) Gating pore current in an inherited ion channelopathy. Nature, 446 (7131): 76-8. [PMID:17330043]
52. Terlau H, Stühmer W. (1998) Structure and function of voltage-gated ion channels. Naturwissenschaften, 85 (9): 437-44. [PMID:9802045]
53. Tikhonov DB, Zhorov BS. (2012) Architecture and pore block of eukaryotic voltage-gated sodium channels in view of NavAb bacterial sodium channel structure. Mol Pharmacol, 82 (1): 97-104. [PMID:22505150]
54. Vargas E, Yarov-Yarovoy V, Khalili-Araghi F, Catterall WA, Klein ML, Tarek M, Lindahl E, Schulten K, Perozo E, Bezanilla F et al.. (2012) An emerging consensus on voltage-dependent gating from computational modeling and molecular dynamics simulations. J Gen Physiol, 140 (6): 587-94. [PMID:23183694]
55. Venance SL, Cannon SC, Fialho D, Fontaine B, Hanna MG, Ptacek LJ, Tristani-Firouzi M, Tawil R, Griggs RC, CINCH investigators. (2006) The primary periodic paralyses: diagnosis, pathogenesis and treatment. Brain, 129 (Pt 1): 8-17. [PMID:16195244]
56. Wang C, Chung BC, Yan H, Lee SY, Pitt GS. (2012) Crystal structure of the ternary complex of a NaV C-terminal domain, a fibroblast growth factor homologous factor, and calmodulin. Structure, 20 (7): 1167-76. [PMID:22705208]
57. Wang GK, Quan C, Wang S. (1998) A common local anesthetic receptor for benzocaine and etidocaine in voltage-gated mu1 Na+ channels. Pflugers Arch, 435 (2): 293-302. [PMID:9382945]
58. Waxman SG, Merkies IS, Gerrits MM, Dib-Hajj SD, Lauria G, Cox JJ, Wood JN, Woods CG, Drenth JP, Faber CG. (2014) Sodium channel genes in pain-related disorders: phenotype-genotype associations and recommendations for clinical use. Lancet Neurol, 13 (11): 1152-60. [PMID:25316021]
59. Yarov-Yarovoy V, Brown J, Sharp EM, Clare JJ, Scheuer T, Catterall WA. (2001) Molecular determinants of voltage-dependent gating and binding of pore-blocking drugs in transmembrane segment IIIS6 of the Na(+) channel alpha subunit. J Biol Chem, 276 (1): 20-7. [PMID:11024055]
60. Yarov-Yarovoy V, DeCaen PG, Westenbroek RE, Pan CY, Scheuer T, Baker D, Catterall WA. (2012) Structural basis for gating charge movement in the voltage sensor of a sodium channel. Proc Natl Acad Sci USA, 109 (2): E93-102. [PMID:22160714]
61. Yarov-Yarovoy V, McPhee JC, Idsvoog D, Pate C, Scheuer T, Catterall WA. (2002) Role of amino acid residues in transmembrane segments IS6 and IIS6 of the Na+ channel alpha subunit in voltage-dependent gating and drug block. J Biol Chem, 277 (38): 35393-401. [PMID:12130650]
62. Yu FH, Catterall WA. (2004) The VGL-chanome: a protein superfamily specialized for electrical signaling and ionic homeostasis. Sci STKE, 2004 (253): re15. [PMID:15467096]
63. Yu FH, Westenbroek RE, Silos-Santiago I, McCormick KA, Lawson D, Ge P, Ferriera H, Lilly J, DiStefano PS, Catterall WA et al.. (2003) Sodium channel beta4, a new disulfide-linked auxiliary subunit with similarity to beta2. J Neurosci, 23 (20): 7577-85. [PMID:12930796]
64. Zhang X, Ren W, DeCaen P, Yan C, Tao X, Tang L, Wang J, Hasegawa K, Kumasaka T, He J et al.. (2012) Crystal structure of an orthologue of the NaChBac voltage-gated sodium channel. Nature, 486 (7401): 130-4. [PMID:22678295]




![]()
 This work is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License
This work is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License