Kinases (EC 2.7.x.x): Introduction

Reversible phosphorylation, where kinases transfer the gamma phosphate of ATP to hydroxyl groups of various substrates including lipids, sugars or amino acids plays an essential role in most, if not all, cellular signaling [32-33], influencing almost every cellular process (including regulation of the cell cycle, growth, apoptosis and signal transduction) [2,5,32]. In eukaryotes, reversible protein phosphorylation is often transient and is performed by ePKs on Ser, Thr or Tyr residues and the respective phosphatases, which remove the phosphate.
At the latest count, the human kinome contains 566 ePK genes. There are two main classes of human protein kinases: the protein tyrosine kinases (PTKs) which phosphorylate Tyr and the Ser- and Thr-specific kinases (STKs) which phosphorylate Ser and/or Thr residues on protein substrates [5-6,32]. Whilst there are a few dual-specificity protein kinases (phosphorylating both Tyr and Thr), the majority of ePKs are STKs. This is reflected in the ratio of cellular phosphorylation of pSer : pThr : pTyr = 1000 : 100 : 1 [2,5-6,16,18,25]. Although only a minor number of substrates are phosphorylated by PTKs, the importance of tyrosine phosphorylation is profound [2,6,12,16,18,25]. Many gain of function (GOF) and/or loss of function (LOF) mutations are found in PTKs which have, therefore, long been considered as important drug targets [2,5-6,12-13,18,21].
The ePK domain is the most abundant catalytic domain in the eukaryotic genome and the overall structural organization of the kinase domain is highly conserved [25]. The catalytic function of ePKs is confined to a ~300 amino acid domain, which provides all of the machinery required for phosphorylation of proteins on Ser, Thr and/or Tyr residues [12,19-20,25,27,29-30,34]. Of special note is the so-called "gatekeeper" residue which controls access to the "back-pocket" of the kinase ATP binding site, [9,20,27,29-30] since this residue is often mutated in kinase alleles resistant to inhibitors.
Aberrant kinase function, such as hyperactivity or overexpression plays a role in a wide variety of diseases including cancer, inflammatory diseases, diabetes, atherosclerosis, and immunological disorders [2,4-5,12,28]. Despite a third of all protein targets under investigation in the pharmaceutical industry being protein or lipid kinases, their full potential remains to be fully exploited [13].
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and the host kinome
In late 2019-early 2020 SARS-CoV-2 infection (COVID-19) swept across the world. The coronavirus has huge effects on host cell biological processes, many of which are regulated by kinase signalling pathways. Bouhaddou et al. (2020) performed quantitative mass spectrometry-based phosphoproteomic analysis of SARS-CoV-2 infected cells to determine how the virus changes and rewires phosphorylation-based signalling, and whether this could provide clues for novel therapeutic intervention points [3]. Key infection-mediated changes that the team uncovered included activation of the host p38 MAP kinase cascade, shutdown of mitotic kinases and identification of casein kinase II-containing filopodia outgrowths that contained budding virus. They were also able to identify potential phosphorylation sites within viral proteins, and host kinase families that could be predicted to regulate these sites and which by inference could regulate viral replication. The pattern of changes in kinase activities was strongly influenced by the stage of viral life cycle (e.g. during viral entry compared to late replication/egress). Of the dysregulated kinases and potential virus protein regulating kinases, pharmacologic inhibition of p38, CK2, CDKs, AXL and PIKFYVE produced in vitro antiviral activity.
KINASE INHIBITORS
Potent but non-selective tool molecules such as staurosporinestaurosporine led to the initiation of drug development efforts which culminated in the approval of the first kinase inhibitor, Imatinibimatinib (CGP57148, STI571, Glivec, Gleevec), for the treatment of chronic myelogenous leukemia (CML) in 2001. Since then, ~50 small molecule kinase inhibitors have been approved by the FDA, the majority being for the treatment of cancer with a minor number approved for other indications [22]. The figure below charts the last 32 years of kinase inhibitor drug discovery, and provides a timeline of kinase inhibitor drug approvals.
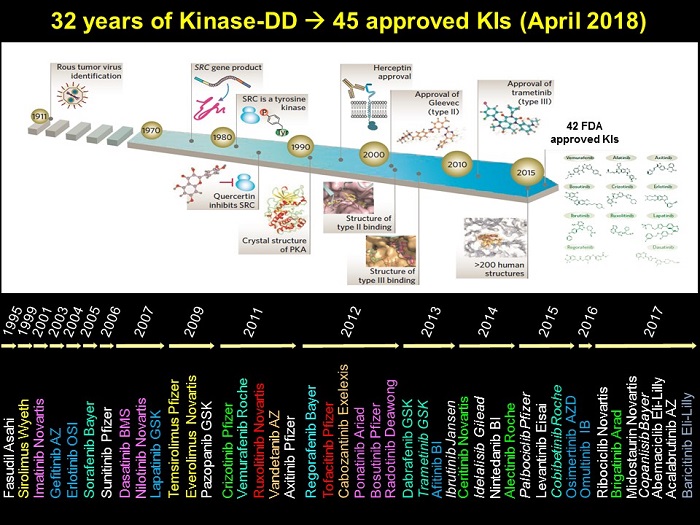
Although the structural determinants of kinase inhibition by small molecules binding to the ATP-binding site is well understood [8,10,12,23,31,34], the selectivity and the limited set of chemical entities targeting the ATP-binding site, have become the major issues in kinase drug discovery.
Traditionally, drug discovery has used in vitro biochemical- and cellular assays followed by in vivo clinical studies to assess drug efficacy. There are many different ways to measure biochemical protein kinase activity (such as detection of radio labeled transfer of phosphate to the substrate, ATP consumption or ADP production measurement, TR-FRET, peptide array-based, micro fluidic technologies, and label free analysis). Cellular assays show similar diversity, focused on measuring target modification (by Western blot, phospho-ELISA and reversed-phase arrays, for example) or changes in downstream signaling. Combining these various techniques gives a readout of in vitro kinase activity and the effect of inhibitors, and can be used to reveal on- and off-target effects. Such systematic inhibitor profiling has provided novel ways to better define the selectivity profile of drug candidates and has revealed previously unidentified mechanisms of action [1,26]. But, despite this enhanced profiling of drug candidates, it still proves very difficult to align these results with their potential toxological effects and clinical efficacy in patients, meaning that many potential research leads fail in pre-clinical stages of development. Recent progress in molecular profiling in conjunction with precision medicine will further our understanding and allow better assessment and prediction of efficacy/toxicity of these inhibitors in disease models (pharmacokinetics/pharmacodynamics) and patients [7,12,15,24,34]. The prospect of using kinase inhibitors against diseases other than cancer may also be enhanced by such improved understanding [14].
Agents other than small molecule compounds are being developed to target kinases.
Monoclonal antibodies (mAbs) and antibody-drug conjugates (ADCs)
The table below lists some of the earliest approved receptor tyrosine kinase mAbs, and indicates target specificity and clinical indications for which the mAbs can be used.
| mAbs | Trade name | Target | Indication |
|---|---|---|---|
| Ramucirumab (Cyramza) | Cyramza | VEGFR-2 | Advanced/metastatic gastric or gastroesophageal junction adenocarcinoma; metastatic non-small cell lung cancer; metastatic colorectal cancer |
| Panitumumab (Vectibix) | Vectibix | EGFR | EGFR-expressing colorectal cancer |
| Pertuzumab (Perjeta) | Perjeta | HER2 | Metastatic HER2-overexpressing breast cancer |
| Cetuximab (Erbitux) | Erbitux | EGFR | Metastatic KRAS negative colorectal cancer; squamous cell carcinoma of the head and neck |
| Trastuzumab | Herceptin, Herclon | HER2 | HER2/neu overexpressing breast cancer, some gastric adenocarcinomas |
| ADC | |||
| Ado-trastuzumab emtansine | Kadcyla | HER2 | Metastatic HER2 over-expressing breast cancer |
Kinase degraders
Proteolysis targeting chimeras (a.k.a. PROTACs) are dual domain molecules that direct targeted proteins towards the intracellular proteolytic degradation pathway. So in essence these agents reduce the physical amount of target protein present, rather than inhibiting its activity. PROTAC degraders work by recruiting an E3 ligase to the unwanted target protein, and this is then recognised and removed by the cells' ubiquitin/proteasome machinery. Degraders can be used against any disease-causing proteins. One kinase specific example is the CDK8 degrader JH-XI-10-02 (which in this case is also a CDK8 inhibitor) [17]. JH-XI-10-02 is a CDK8 inhibitor/pomalidomide chimeric molecule that binds to the ubiquitously expressed E3 ligase receptor cereblon via pomalidomide. JH-XI-10-02 thereby recruits E3 ligase CRL4cereblon to inhibitor-bound CDK8 and directs its destruction via cereblon-mediated ubiquitination and proteosomal degradation.
References
1. Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D et al.. (2012) The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature, 483 (7391): 603-7. [PMID:22460905]
2. Blume-Jensen P, Hunter T. (2001) Oncogenic kinase signalling. Nature, 411 (6835): 355-65. [PMID:11357143]
3. Bouhaddou M, Memon D, Meyer B, White KM, Rezelj VV, Marrero MC, Polacco BJ, Melnyk JE, Ulferts S, Kaake RM. (2020) The Global Phosphorylation Landscape of SARS-CoV-2 Infection. Cell, Article Online Now. DOI: 10.1016/j.cell.2020.06.034
4. Chico LK, Van Eldik LJ, Watterson DM. (2009) Targeting protein kinases in central nervous system disorders. Nat Rev Drug Discov, 8 (11): 892-909. [PMID:19876042]
5. Cohen P. (2001) The role of protein phosphorylation in human health and disease. The Sir Hans Krebs Medal Lecture. Eur J Biochem, 268 (19): 5001-10. [PMID:11589691]
6. Cohen P. (2002) Protein kinases--the major drug targets of the twenty-first century?. Nat Rev Drug Discov, 1 (4): 309-15. [PMID:12120282]
7. Courtney KD, Corcoran RB, Engelman JA. (2010) The PI3K pathway as drug target in human cancer. J Clin Oncol, 28 (6): 1075-83. [PMID:20085938]
8. Cowan-Jacob SW. (2006) Structural biology of protein tyrosine kinases. Cell Mol Life Sci, 63 (22): 2608-25. [PMID:17041812]
9. Cowan-Jacob SW, Möbitz H, Fabbro D. (2009) Structural biology contributions to tyrosine kinase drug discovery. Curr Opin Cell Biol, 21 (2): 280-7. [PMID:19208462]
10. Engelman JA. (2009) Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer, 9 (8): 550-62. [PMID:19629070]
11. Fabbro D, Cowan-Jacob SW, Moebitz H. (2015) Ten things you should know about protein kinases: IUPHAR Review 14. Br J Pharmacol, 172 (11): 2675-700. [PMID:25630872]
12. Fabbro D, Cowan-Jacob SW, Möbitz H, Martiny-Baron G. (2012) Targeting cancer with small-molecular-weight kinase inhibitors. Methods Mol Biol, 795: 1-34. [PMID:21960212]
13. Fedorov O, Müller S, Knapp S. (2010) The (un)targeted cancer kinome. Nat Chem Biol, 6 (3): 166-169. [PMID:20154661]
14. Ferguson FM, Gray NS. (2018) Kinase inhibitors: the road ahead. Nat Rev Drug Discov, 17 (5): 353-377. DOI: 10.1038/nrd.2018.21 [PMID:29545548]
15. Gray-Schopfer V, Wellbrock C, Marais R. (2007) Melanoma biology and new targeted therapy. Nature, 445 (7130): 851-7. [PMID:17314971]
16. Hanks SK, Hunter T. (1995) Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J, 9 (8): 576-96. [PMID:7768349]
17. Hatcher JM, Wang ES, Johannessen L, Kwiatkowski N, Sim T, Gray NS. (2018) Development of Highly Potent and Selective Steroidal Inhibitors and Degraders of CDK8. ACS Med Chem Lett, 9 (6): 540-545. [PMID:29937979]
18. Hunter T. (2000) Signaling--2000 and beyond. Cell, 100 (1): 113-27. [PMID:10647936]
19. Kannan N, Taylor SS, Zhai Y, Venter JC, Manning G. (2007) Structural and functional diversity of the microbial kinome. PLoS Biol, 5 (3): e17. [PMID:17355172]
20. Kornev AP, Haste NM, Taylor SS, Eyck LF. (2006) Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc Natl Acad Sci USA, 103 (47): 17783-8. [PMID:17095602]
21. Lahiry P, Torkamani A, Schork NJ, Hegele RA. (2010) Kinase mutations in human disease: interpreting genotype-phenotype relationships. Nat Rev Genet, 11 (1): 60-74. [PMID:20019687]
22. Lightfoot HL, Goldberg FW, Sedelmeier J. (2019) Evolution of Small Molecule Kinase Drugs. ACS Med Chem Lett, 10 (2): 153-160. DOI: 10.1021/acsmedchemlett.8b00445 [PMID:30783496]
23. Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH, Gray NS. (2013) Developing irreversible inhibitors of the protein kinase cysteinome. Chem Biol, 20 (2): 146-59. [PMID:23438744]
24. London CA. (2013) Kinase dysfunction and kinase inhibitors. Vet Dermatol, 24 (1): 181-7.e39-40. [PMID:23331696]
25. Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. (2002) The protein kinase complement of the human genome. Science, 298 (5600): 1912-34. [PMID:12471243]
26. Melnick JS, Janes J, Kim S, Chang JY, Sipes DG, Gunderson D, Jarnes L, Matzen JT, Garcia ME, Hood TL et al.. (2006) An efficient rapid system for profiling the cellular activities of molecular libraries. Proc Natl Acad Sci USA, 103 (9): 3153-8. [PMID:16492761]
27. Moebitz H, Fabbro D. (2012) Conformational bias: A key concept for protein kinase inhibition. European Pharmaceutical Review, 17: 41-51.
28. Müller S, Knapp S. (2010) Targeting kinases for the treatment of inflammatory diseases. Expert Opin Drug Discov, 5 (9): 867-81. [PMID:22823261]
29. Nolen B, Taylor S, Ghosh G. (2004) Regulation of protein kinases; controlling activity through activation segment conformation. Mol Cell, 15 (5): 661-75. [PMID:15350212]
30. Taylor SS, Kornev AP. (2011) Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem Sci, 36 (2): 65-77. [PMID:20971646]
31. Traxler P, Bold G, Buchdunger E, Caravatti G, Furet P, Manley P, O'Reilly T, Wood J, Zimmermann J. (2001) Tyrosine kinase inhibitors: from rational design to clinical trials. Med Res Rev, 21 (6): 499-512. [PMID:11607931]
32. Ubersax JA, Ferrell JE. (2007) Mechanisms of specificity in protein phosphorylation. Nat Rev Mol Cell Biol, 8 (7): 530-41. [PMID:17585314]
33. Walsh CT, Garneau-Tsodikova S, Gatto Jr GJ. (2005) Protein posttranslational modifications: the chemistry of proteome diversifications. Angew Chem Int Ed Engl, 44 (45): 7342-72. [PMID:16267872]
34. Zhang J, Yang PL, Gray NS. (2009) Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer, 9 (1): 28-39. [PMID:19104514]
How to cite this page
To cite this family introduction, please use the following:
Kinases (EC 2.7.x.x), introduction. Last modified on 15/02/2021. Accessed on 20/04/2024. IUPHAR/BPS Guide to PHARMACOLOGY, https://www.guidetoimmunopharmacology.org/GRAC/FamilyIntroductionForward?familyId=698.





![]()
 This work is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License
This work is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License